
 Because viruses must bind to cell surface molecules to initiate replication, the use of soluble receptors to block virus infection has long been an attractive therapeutic option. Soluble receptors have been developed that block infection with rhinoviruses and HIV-1, but these have not been licensed due to their suboptimal potency. A newly designed soluble receptor for HIV-1 overcomes this problem and provides broad and effective protection against infection of cells and of nonhuman primates.
Because viruses must bind to cell surface molecules to initiate replication, the use of soluble receptors to block virus infection has long been an attractive therapeutic option. Soluble receptors have been developed that block infection with rhinoviruses and HIV-1, but these have not been licensed due to their suboptimal potency. A newly designed soluble receptor for HIV-1 overcomes this problem and provides broad and effective protection against infection of cells and of nonhuman primates.
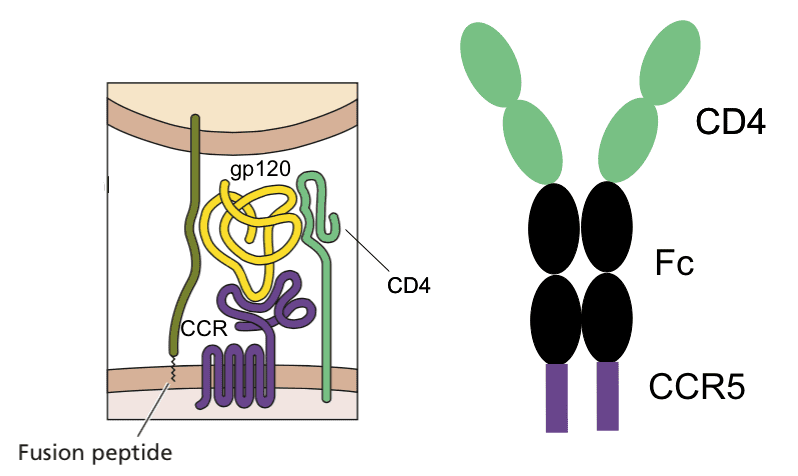
Infection with HIV-1 requires two cell surface molecules, CD4 and a chemokine receptor (either CCR5 or CXCR4), which are engaged by the viral glycoprotein gp120 (illustrated). A soluble form of CD4 fused to an antibody molecule can block infection of most viral isolates, and has been shown to be safe in humans, but its affinity for gp120 is low. Similarly, peptide mimics of the CCR5 co-receptor have been shown to block infection, but their affinity for gp120 is also low.
Combining the two gp120-binding molecules solved the problem of low affinity, and in addition provided protection against a wide range of virus isolates. The entry inhibitor, called eCD4-Ig, is a fusion of the first two domains of CD4 to the Fc domain of an antibody molecule, with the CCR5-mimicking peptide at the carboxy-terminus (illustrated). It binds strongly to gp120, and blocks infection with many different isolates of HIV-1, HIV-2, SIV, and HIV-1 resistant to broadly neutralizing monoclonal antibodies. The molecule blocks viral infection at concentrations that might be achieved in humans (1.5 – 5.2 micrograms per milliliter).
When administered to mice, eCD4-Ig protected the animals from HIV-1. Rhesus macaques inoculated with an adenovirus-associated virus (AAV) recombinant containing the gene for eCD4-Ig were protected from infection with large amounts of virus for up to 34 weeks after immunization. Levels of eCD4-Ig in the sera of these animals ranged from 17 – 77 micrograms per milliliter.
These results show that eCD4-Ig blocks HIV infection with a wide range of isolates more effectively than previously studied broadly neutralizing antibodies. Emergence of HIV variants resistant to neutralization with eCD4-Ig would likely produce viruses that infect cells less efficiently, reducing their transmission. eCD4-Ig is therefore an attractive candidate for therapy of HIV-1 infections. Whether sustained production of the protein in humans will cause disease remains to be determined. Because expression of the AAV genome persists for long periods, it might be advantageous to include a kill-switch in the vector: a way of turning it off if something should go wrong.
Dumping soluble CD4 into the circulation is just going screw up APCs and probably lead to a slew of autoimmune problems. There are definitely better approaches to take than this.
My guess is that the CD4/Fc/CCR5mim1 complexes have low affinity for MHC class II. If that’s not the case, the CD4 domains could probably be modified to still be able to neutralize the virus but not bind to MHC class II.
What do you think is a better approach?
Pingback: Blocking virus infection with soluble cell receptors