
by Gertrud U. Rey
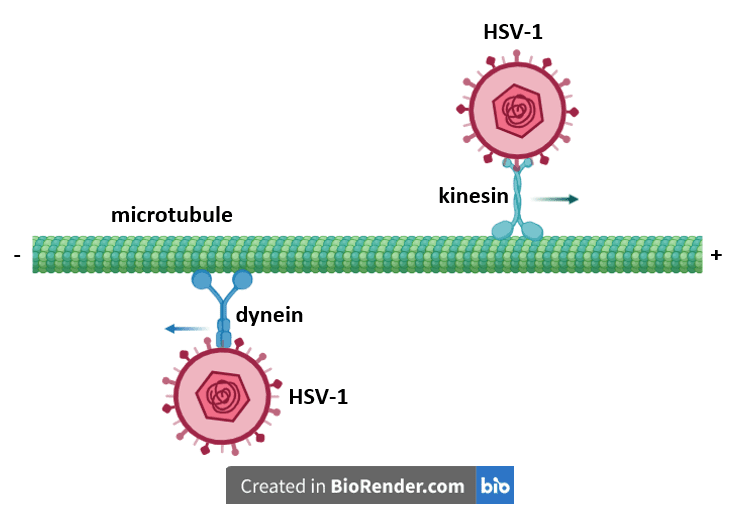
Viruses that require access to the host cell nucleus typically get there by exploiting cellular motor proteins for their own transportation purposes. For example, herpesviruses use two cellular proteins called dynein and kinesin to shuttle their cargo to and from the nucleus. However, the exact mechanism by which these viruses engage the motor proteins was unclear until recently.
Dynein and kinesin traffic around the cell on long cylindrical structures called microtubules, which not only serve as a “railway” system for a variety of motor proteins that transport cargo to support various cellular functions, but also provide a framework for the cytoskeleton. Microtubules have a well-defined polarity that is integral to their biological function: one end is designated the plus end, while the other end is the minus end. Dynein moves from the plus end of the microtubule towards the minus end in the direction of the nucleus, while kinesin moves in the opposite direction, typically leading to the cell periphery.
Herpes simplex virus type 1 (HSV-1) initially infects epithelial cells of the skin and mucous membranes. During this infection, viral particles fuse with the host cell membrane and release the protein shell that encompasses the viral genome (the “capsid”), into the cytoplasm. A virally encoded and capsid-bound protein called pUL36 then binds dynein, allowing the capsid to travel along a microtubule to the nucleus, where the viral genome is injected through a nuclear pore. When newly assembled capsids carrying newly replicated viral genomes exit from the nucleus at a later time, they engage kinesin to motor along the microtubules toward the cell membrane. Newly formed virions emerging from the cell membrane subsequently infect nearby sensory neurons, where they establish a lifelong, persistent infection.
Studies have shown that deletion of the portion of pUL36 that tethers it to the viral capsid results in its ability to move along microtubules in both directions, suggesting that in addition to binding dynein, pUL36 also binds kinesin. Subsequent follow-up studies exploring the kinetics of this presumed interaction revealed that pUL36 does indeed bind kinesin through a series of tryptophan-aspartate (WD) motifs. Two of these motifs – WD3 and WD4 – appear to be particularly important: WD3 improves pUL36-kinesin binding and WD4 is essential for the interaction.
In an effort to more clearly define the role of kinesin during herpesvirus infection, Caitlin Pegg, Sofia Zaichick and co-authors of a recent study, carried out a series of experiments using HSV-1 and pseudorabiesvirus (PRV), a herpesvirus in the same subfamily as HSV-1, which is often used as a model to study the basic processes of herpesvirus infection. To determine whether WD3 and WD4 are needed for PRV replication and viability, the authors introduced mutations in the DNA sequences encoding these motifs in PRV. Plaque assays done using cells infected with the respective modified viruses revealed that viruses with a mutation in WD3 produced smaller plaques, suggesting that this motif enhanced viral replication. Viruses with a mutation in WD4 produced almost no plaques, suggesting that this motif was essential for viral replication. These results complemented the data obtained from the pUL36-kinesin binding studies, implying that an interaction between pUL36 and kinesin is needed for viral replication.
The authors also determined that a mutation in the sequence encoding the acidic amino acids of the WD4 motif led to reduced kinesin binding but did not impair PRV replication. To study the transport mechanism of PRV, they generated a virus with reduced kinesin binding ability (“RKB”) by changing the amino acids in both WD3 and the acidic amino acids in WD4. They further modified the virus by inserting a gene encoding a fluorescent marker next to the capsid genes. The joint expression of the capsid and marker genes in cells would result in fluorescently tagged capsids, allowing one to visualize these particles using a fluorescence microscope. Microscopic analysis of sensory neurons infected with fluorescently tagged RKB viruses revealed that the capsids traveled through the cytoplasm toward the nucleus, but never actually reached the nucleus. This suggests that pUL36 binding to kinesin is important for delivery of viral particles into the neuronal nucleus during infection.
To further substantiate this finding, the authors performed infection experiments using retinal pigment epithelial (RPE) cells that do not express kinesin. Infection of these cells with fluorescently-tagged HSV-1 revealed that viral capsids only advanced to the centrosome, an organelle involved in cell division that is somewhat removed from the nucleus. In contrast, infection of wild type RPE cells having functional kinesin resulted in trafficking of viral capsids right up to the nucleus, confirming that kinesin is needed for transporting capsids from the centrosome to the nucleus.
To determine what happens when virions are produced in the absence of kinesin, the authors infected kinesin-deficient RPE cells with either wild type HSV-1 or wild type PRV, isolated the resulting viral particles and then used those viruses to infect wild type (i.e., kinesin-expressing) RPE cells. Visualization of capsid movement in the wild type RPE cells revealed that the capsids only moved as far as the centrosome, and that centrosome-to-nucleus trafficking was impaired, an effect that was particularly pronounced when the experiment was repeated using wild type sensory neurons. The fact that the virus particles isolated from kinesin-deficient cells were unable to traffic to the nucleus in neuronal cells in spite of the fact that these cells produced kinesin suggests that viruses that do make it to the neuronal nucleus must typically carry kinesin when they enter these cells, presumably inside the virion. However, analysis of wild type cell-originating HSV-1 extracellular virions using anti-kinesin antibodies did not reveal the presence of kinesin in these virions.
In a different approach aimed at identifying kinesin inside HSV-1 virions, the authors generated a DNA construct consisting of kinesin fused to the reporter enzyme β-lactamase and inserted this construct into kinesin-deficient RPE cells. They then infected the cells with HSV-1, extracted newly replicated virions from the cells and mixed the virions with nitrocefin, a molecule that reacts with β-lactamase to produce a color change that can be visually detected. A color change would indicate the presence of β-lactamase, which would mean that kinesin was present as well. This method revealed that the virions isolated from the kinesin/β-lactamase-expressing cells did contain β-lactamase, further suggesting that they also contained kinesin. These results suggested that HSV-1 virions capture kinesin from initially infected epithelial cells and ferry it to subsequently infected neuronal cells to deliver their genome into the nucleus of those cells.
To confirm whether this hypothesis was true, the authors prepared HSV-1 virions expressing kinesin fused to DmrB, a protein that can be induced to dimerize through addition of a drug. When the drug was added to primary sensory neurons infected with HSV-1 carrying the DmrB-kinesin fusion protein, capsid transport from the centrosome to the nucleus was reduced, presumably because DmrB formed a dimer which structurally impaired the motility of kinesin. These results confirmed that kinesin mediates the movement of capsids from the centrosome to the nucleus. And although kinesin-mediated transport of HSV-1 presumably leads the virions to the cell membrane instead of the nucleus, the microtubules radiating out from the centrosome toward the nucleus are oriented such that their minus end is anchored in the centrosome, while the plus end is at the nucleus, consistent with the directional movement of kinesin.
The fact that viruses can capture a cellular protein and incorporate it for later use is a remarkable discovery that the authors refer to as “assimilation.” As more research emerges in this field, it will be interesting to see whether herpesviruses steal other cellular proteins and whether members of other virus families also assimilate cellular proteins to suit their purposes.
[For an in-depth discussion of this paper, please check out TWiV 833. The material in this blog post is also covered in episode 30 of Catch This.]