

Virus particles are metastable: they must be highly stable, to protect the nucleic acid while traveling from host to host or cell to cell, and then come apart to liberate the genome into the cell. New insight into this uncoating process comes from beautiful images of feline calicivirus showing formation of a portal on the capsid upon receptor engagement.
How virus particles become destabilized has been the subject of much research interest for the past 20 years, a field that has been enhanced by high resolution x-ray crystallographic and cryo-electron microscopy structural data. One solution for viruses with protein shells built with icosahedral symmetry is to release the genome through a portal in the capsid. Examples include herpes simplex virus, where a single portal is the site of nucleic acid entry and exit; and picornaviruses, where a portal appears to be induced by receptor engagement.
Caliciviruses are small viruses with RNA genomes; one member, norovirus, is well known for causing gastroenteritis in humans. The virus particles are built with 180 copies of the capsid protein VP1 in a t=3 icosahedral shell (to understand virus structure, watch my video). A minor but essential capsid protein, VP2, is also present.
Feline calicivirus initiates infection by binding to the cell receptor fJAM-A. Visualization of the structure of the virus bound to fJAM-A by cryo-electron microscopy revealed a single portal at one three-fold axis of symmetry – a structure not seen in calicivirus without receptor added (image above). Twelve molecules of VP2 can be seen forming the portal; at its base is an opening in the capsid shell.
Below is a side view of the portal; the 12 extended VP2 molecules (red) are clearly visible.
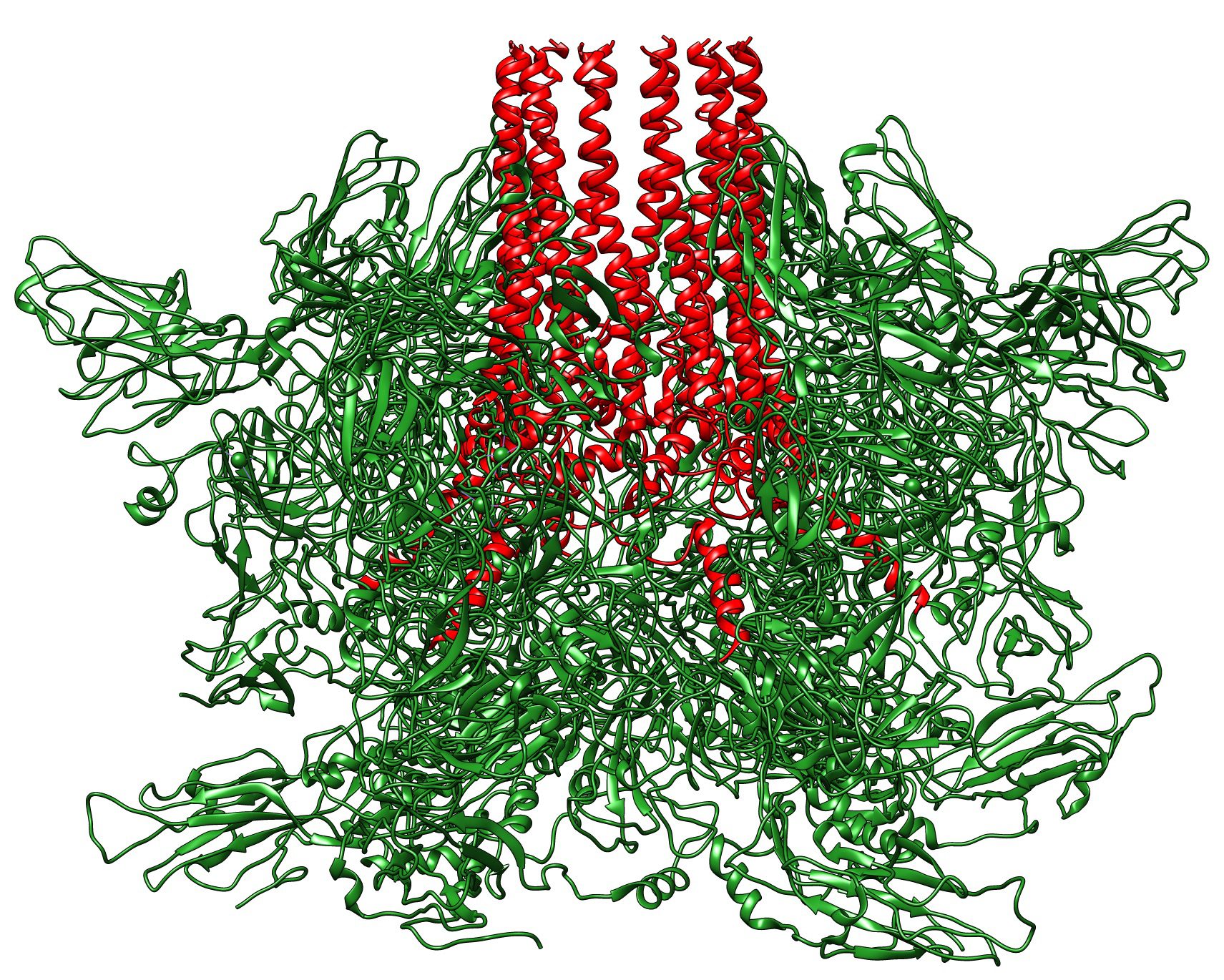
These structures suggest the following scenario for calicivirus entry into cells. In the native capsid, 12 molecules of VP2 are buried at one two-fold axis of symmetry. Upon binding fJAM-A on the cell surface, VP1 and VP2 undergo conformational changes that extend VP2, forming the portal and the hole in the capsid. The amino termini of VP2 are hydrophobic, and likely penetrate the cell membrane as the virus particle is taken up by endocytosis. The viral RNA then passes out of the capsid, through the portal and the membrane channel formed by the amino-termini of VP2.
It makes sense that the amino-termini of VP2 are buried in the capsid before receptor engagement: they need to be hidden so that fusion with the wrong cell will not occur. Only upon receptor engagement – signifying a potentially ‘correct€™ cell for replication – are the amino-termini of VP2 exposed. Similar strategies for regulating membrane fusion can be found in many virus reproduction pathways.
The hole in the capsid below the portal is about 1 nanometer in diameter – enough to allow an RNA to thread through. As there is only one portal on the viral capsid, this observation begs the question of how the RNA end is oriented so that it is positioned below the portal, ready for release. This arrangement – RNA end below the portal – must be established during virus assembly. If the RNA end were randomly arranged in the capsid, the likelihood that it would be below the portal would be slim, greatly reducing viral infectivity. How this assembly takes place will be important to establish.
Ever since my laboratory identified the cell receptor for poliovirus back in 1989, I have been interested in understanding how the receptor caused viral RNA to leave the capsid. That’s why the finding of a unique calicivirus portal for RNA exit is particularly engaging for me!
For more discussion of the calicivirus portal, listen to This Week in Virology #534.
Pingback: A portal for RNA exit – Virology Hub
Pingback: A portal for RNA exit -