
 All immune responses, from intrinsic to adaptive, need to be regulated – if left on indefinitely they will damage the host. The innate immune response is no exception, and a cellular RNA has been identified that binds to a sensor of viral RNA and regulates the production of interferon (IFN).
All immune responses, from intrinsic to adaptive, need to be regulated – if left on indefinitely they will damage the host. The innate immune response is no exception, and a cellular RNA has been identified that binds to a sensor of viral RNA and regulates the production of interferon (IFN).
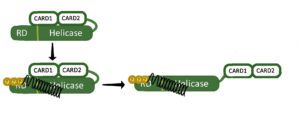
The innate immune response, an early response to viral infection, depends on sensors that can distinguish self from non-self. One sensor of non-self RNA is RIG-I, a cytoplasmic protein that binds to short, blunt ended dsRNAs containing a 5€²-triphosphate. Such RNAs are not typically found in the cell cytoplasm, and hence are a signature of virus infection. The RIG-I protein contains two N-terminal caspase recruitment domains (CARDs), a central helicase/ATPase domain, and a C-terminal regulatory domain (RD) (Figure). In an uninfected cell, the CARD domains are blocked by interaction with other parts of the protein (Figure). When RNA binds RIG-I (black spring in figure) it induces a conformational change, exposing the CARDs. The released CARDs bind MAVS, a mitochondrial outer membrane protein, to start a signaling cascade that leads to the synthesis of IFN.
A long non-coding RNA called Lsm3b has been identified in mouse cells that binds RIG-I but does not lead to the synthesis of IFN (link to paper). Instead, Lsm3b dampens IFN production late in RNA virus-infected cells. When Lsm3b is removed, IFN production increases.
Lsm3b synthesis is induced by IFN, but only after IFN is produced after 12-16 hours. Then levels of Lsm3b rise to sufficient levels to shut down IFN synthesis.
Lsm3b RNA is a competitive inhibitor of viral RNA binding to RIG-I. Furthermore, its mechanism of binding is completely different from that of viral RNA, and it binds with greater affinity. Binding of Lsm3b to RIG-I does not lead to exposure of the CARD domains (Figure) and therefore does not induce IFN synthesis.
As Lsm3b levels rise late in infection, the RNAs bind up all the free RIG-I in cells, shutting down IFN production. This regulation is achieved because IFN is toxic: if cells have survived infection, there is no point in having them killed by IFN!
Lsm3b is a negative regulator of RIG-I signaling during RNA virus infection. Immune homeostasis is therefore achieved by self-recognition of host RNAs.
One can imagine using the human version of Lsm3b to control autoimmune inflammatory diseases. However, human cells do not produce Lsm3b! All the work described above was done in mouse cells. Perhaps human cells produce a different lncRNA, or maybe utilize a completely different mechanism to regulate RIG-I activation.
Pingback: A cell RNA that regulates innate immunity – Virology
Pingback: A cell RNA that regulates innate immunity - Vetmedics
Pingback: A cell RNA that regulates innate immunity -