
By Gertrud U. Rey
Gertrud Rey is a trained virologist residing in Atlanta, Georgia. During the day, she works as a consultant in a biotech patent law firm, but spends much of her free time as a science communicator. She was a guest on TWiV 179 and 424.
The lack of a suitable animal model for human dengue virus infection and disease has presented considerable challenges for dengue virus vaccine research.
Chimpanzees, rhesus macaques, and the common marmoset, representing apes, Old World monkeys, and New World monkeys, respectively, have been used as model organisms to study dengue. However, although they are permissive for dengue virus infection, they do not develop overt disease. Having good animal models to understand the interaction between dengue virus and the host innate immune response is particularly important for vaccine development.
A key outcome of innate immune responses to viruses is the production of type I interferon. In the innate immune sensing pathway for viruses, the endoplasmic reticulum resident protein STING (“STimulator of INterferon Genes”) plays an important role. As illustrated below, STING is activated by the cytoplasmic sensor cGAS in response to double stranded DNA (dsDNA), whose presence in the cytoplasm is rare and usually indicates infection or cellular damage. Activated STING protects infected cells by mediating the phosphorylation of the transcription factor interferon regulatory factor 3 (IRF3), which in turn induces the synthesis of type I interferon, leading to reduction of viral titers.
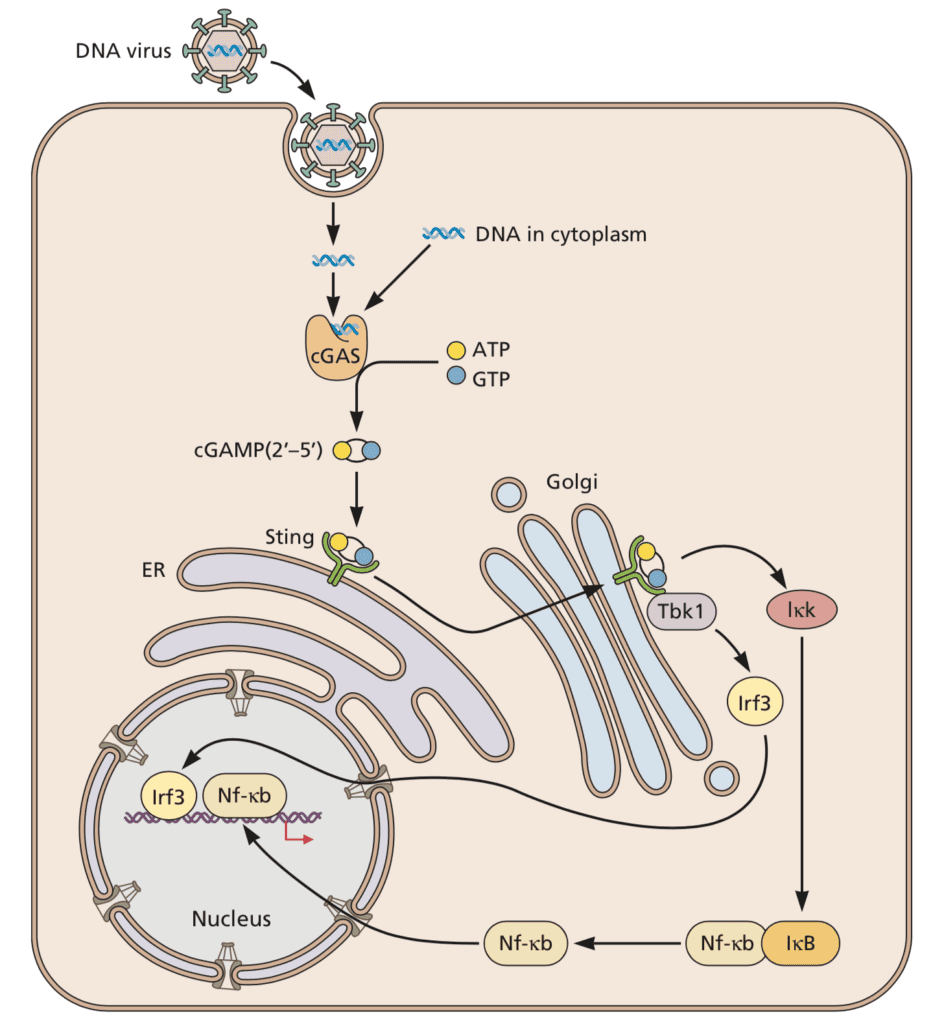
Image credit: Principles of Virology, 4thedition
As described in a previous post, STING is targeted by DNA viruses such as herpesvirus, which inactivates it by binding it and blocking interferon responses to cytoplasmic DNA. Despite its inactivation in response to dsRNA, surprisingly, STING is also inactivated by proteins of several RNA viruses. For example, the dengue virus 2 (DENV2)-encoded protease NS2B3 cleaves STING, thereby preventing the phosphorylation of IRF3 and its downstream effects, and leading to an increase in DENV2 titers. One reason why an RNA virus protease such as that of dengue virus might inactivate the cGAS/STING pathway might be because virus infection damages the mitochondrion, leading to DNA leakage, which is then sensed by cGAS.
In a recent study conducted at the University of Colorado, Alex Stabell and colleagues analyzed the ability of human and sylvatic dengue virus proteases to cleave the STING protein of humans and non-human primates. The term “sylvatic” refers to pathogens that occur in wild animals; the natural reservoir for dengue virus is presently unknown. As pictured below, the DENV NS2B and NS3 protein genes are adjacent to each other in the viral genome and are translated together as one long polyprotein. Following translation, the NS2B-NS3 region cleaves itself from the polyprotein to become the functional protease complex.
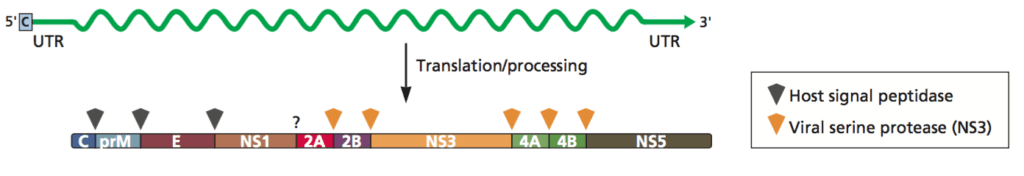
Image credit: Principles of Virology, 4thedition
The authors generated constructs encoding FLAG epitope tag peptides fused to the C-terminus of either wild type (active) or mutant (inactive) DENV2 proteases. They also generated constructs encoding the HA epitope fused to the C-terminus of human, chimp, macaque, marmoset, or mouse STING proteins. Mouse STING is resistant to DENV protease cleavage and served as a negative control. Cells were then co-transfected with plasmids encoding all possible variations of one protease construct and one STING construct. One day later, cell lysates were analyzed by Western blot with antibodies to the FLAG and HA epitopes. Results suggested that the active DENV2 protease, which cleaved human STING efficiently, was unable to cleave any of the other STING proteins of nonhuman primates, including that of chimps. This observation was surprising, considering that human and chimp STING proteins differ at only three amino acid residues: positions 78, 230, and 232.
The next step was to identify the precise STING cleavage site used by DENV2 protease. Previous studies had mapped this cleavage between residues 95 and 96 of STING, a site which is identical in human and chimp STING proteins. However, some doubt remained about the accuracy of this putative cleavage site, because changing the amino acids at residues 95 and 96 in mouse STING to the human sequence did not confer cleavage susceptibility in mice. The authors found that changing the arginine at position 78 in human STING to the tryptophan encoded in chimp STING resulted in resistance to cleavage by DENV NS2B3. The opposite was also true: change of the chimp tryptophan to the human arginine caused the protein to become susceptible to cleavage. In contrast, alteration at residue 230 alone or in combination with residue 78 had no effect. These results suggest that arginine 78 is important for viral cleavage.
Previous studies had suggested that residue 78 is important for retaining STING in the endoplasmic reticulum. To ensure that the introduced mutations in human and chimp STING did not disrupt this retention, the authors generated human STING knockout cells and then stably complemented them with wild type or cleavage resistant human STING. Results showed that both the wild type and mutant STING similarly localized to the ER.
To determine whether cleavage of STING affects its ability to signal in the interferon induction pathway, the authors co-transfected plasmids encoding STING and DENV2 protease into human cells, and two days later, assessed phosphorylation of IRF3 by Western blot. In cases where STING was cleaved, phosphorylation of IRF3 was reduced. They also assessed the activation of the type I interferon beta (IFNb) promoter by co-transfecting cells with plasmids encoding an IFNb/firefly luciferase fusion protein in addition to the usual plasmids encoding STING and NS2B3. The activity of the IFNb promoter was significantly reduced in the presence of active NS2B3 protease. These results were then confirmed with infection experiments. STING knockout cells were complemented with either cleavage-resistant or -susceptible chimp or human forms of STING and infected with DENV2. Cells that were complemented with a cleavage-resistant version of STING produced less virus than those complemented with a susceptible STING, suggesting that cleavage of STING is critical for DENV replication.
To verify that the arginine/glycine (RG) motif at position 78/79 is indeed critical for cleavage by the DENV protease, the authors introduced changes at this site in the STING proteins of rhesus macaque, marmoset, and mouse, all of which are naturally resistant to cleavage. A change to the human RG motif rendered the STING proteins of all of these animals susceptible to cleavage.
A final set of experiments showed that the proteases of the other three endemic human dengue viruses (DENV1, DENV3, and DENV4) and of sylvatic dengue viruses also preferentially cleave the RG motif at position 78/79 of human STING.
A sequence alignment of the region surrounding the RG motif of STING in 30 primate species shows that all apes except chimps and bonobos encode the same motif as humans. Mice and pigs, however, which constitute the two main current models for dengue virus infection, do not encode the correct RG sequence. This observation may be one of the reasons why it has been so difficult to model dengue virus infection in animals. Further query of STING sequences available from placental mammals revealed that the chinchilla, naked mole rat, and desert woodrat all share the same eight amino acid sequence surrounding the RG motif as that found in humans.
Identifying mechanisms for the regulation of viral infection that differ between species is important to our understanding of viral adaptation during host switching. It is also crucial for the identification of appropriate animal models for the study of human viruses. In particular, the finding that genomes of the common chinchilla, naked mole rat, and desert woodrat encode STING proteins susceptible to DENV protease cleavage is noteworthy, especially considering that these species are already used as animal models in biomedical research and that their genome sequences are available. Since these models have an intact STING pathway and presumably maintain the authenticity of signaling induced by viral infection, they could present a clear advantage over STING knockout mice as animal models for DENV infection.