
 Virus populations do not consist of a single member with a defined nucleic acid sequence, but are dynamic distributions of nonidentical but related members called a quasispecies (illustrated at left). While next-generation sequencing methods have the capability of describing a quasispecies, the errors associated with this technology have limited progress in our understanding of the genetic structure of virus populations. A new method called CirSeq reduces next-generation sequencing errors to allow an accurate description of viral quasispecies.
Virus populations do not consist of a single member with a defined nucleic acid sequence, but are dynamic distributions of nonidentical but related members called a quasispecies (illustrated at left). While next-generation sequencing methods have the capability of describing a quasispecies, the errors associated with this technology have limited progress in our understanding of the genetic structure of virus populations. A new method called CirSeq reduces next-generation sequencing errors to allow an accurate description of viral quasispecies.
The key to eliminating sequencing errors is a clever approach based on the conversion of viral RNAs to circular molecules. When copied with reverse transcriptase, tandemly repeated cDNAs are produced (illustrated below). Mutations in the original viral RNA will be shared by all repeats derived from a circle, but not errors produced during copying or sequencing. The latter can be computationally subtracted, reducing sequencing error to a point that is much lower than the estimated mutation rate of an RNA virus.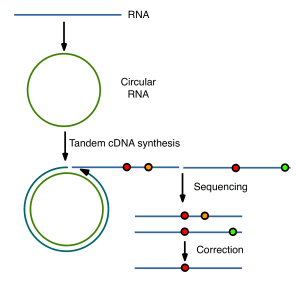
CirSeq was used to characterize poliovirus populations produced by seven serial passages in HeLa cells. The calculated mutation frequency, 2 X 10-4 mutations per nucleotide, was substantially lower compared with estimates determined by conventional sequence analysis. Over 200,000 sequence reads per nucleotide position were used to detect >16,500 variants per population per passage. This number represents ~74% of all possible alleles. Many mutations were detected at nearly all positions in the viral RNA. Most mutations occur at a frequency between 1 in 1000 to 1 in 100,000. The conclusion is that the virus population produced in HeLa cells consists mainly of genomes with the consensus sequence, and small amounts of many variant genomes. These variants are only those that give rise to viable viruses; lethal mutations are not observed.
CirSeq was also used to calculate the mutation rate of poliovirus. The rates vary according to type: transitions occurred at a rate of 2.5 X 10-5 to 2.6 X 10-4 substitutions per site, while transversions were observed at a rate of 1.2 X 10-6 to 1.5 X 10-5 substitutions per site. Nucleotide-specific differences in mutation rate were also observed: C to U and G to A transitions were 10 times more frequent than U to C and A to G. These rates are consistent with previously determined values using other methods.
This method can also be used to determine the fitness of each base at every position in the genome, according to changes observed during the seven passages in HeLa cells. This analysis allows determination of which bases are neutral, and which are selected, and when combined with analysis of protein structure, can provide new insights into viral functions.
By enabling a sequencing approach that gives an accurate description of virus populations at a single-nucleotide level, CirSeq can be used to provide an unprecedented view of how virus populations change during evolution.
Nice
I really like the idea of Cirseq, and I am considering to apply it on my insect virus populations. However, I wonder whether doing more in depth paired-end RNA sequencing (higher coverage) would able to get the same precision without having to circularize the cDNA library prior sequencing? And for the same cost.
It’s true that sequencing at high depth might be able to diminish the impact of background mutations (from PCR and/or sequencing). For example, you could intentionally target an average of 20 read pairs for every 1 starting biological molecule, and then eliminate/mask mutations that are observed less than 10 times (as these would be mostly background errors). However, there are several limitations of doing this. First, you likely wouldn’t be able to distinguish biological mutations from early PCR errors. Second, some background errors are unfortunately non-random..i.e. biased toward certain types and positions. So even if you applied some sort of cutoff, as I listed above, you would still obtain numerous background errors above the threshold, although these could potentially be identified and masked using some sort of control to measure background errors. Third, this would require extensive re-sequencing, such that you might obtain the same number of reads off the sequencer as in CirSeq, but you would be sampling far fewer variants.
One could also avoid circularizing and many spurious errors by sequencing replicate libraries from the same RNA prep.
Now computational data analysis of CirSeq data possible on T-Bioinfo: http://pine-biotech.com/industry-type/virology/