
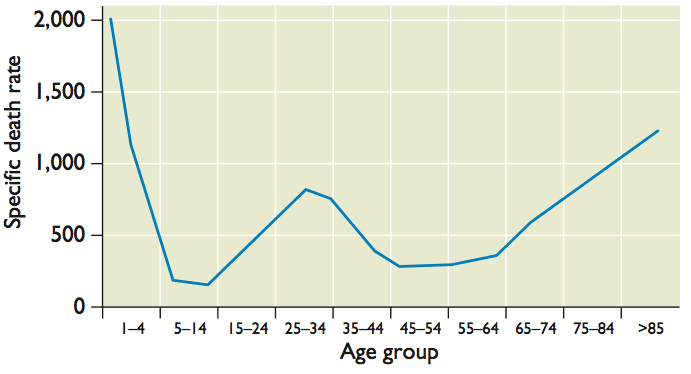
 The 1918 influenza pandemic was particularly lethal, not only for the very young and the very old (as observed for typical influenza), but unexpectedly also for young adults, 20 to 40 years of age (pictured). It has been suggested that the increased lethality in young adults occurred because they lacked protective immunity that would be conferred by previous infection with a related virus. Reconstruction of the origins of the 1918 influenza virus provides support for this hypothesis.
The 1918 influenza pandemic was particularly lethal, not only for the very young and the very old (as observed for typical influenza), but unexpectedly also for young adults, 20 to 40 years of age (pictured). It has been suggested that the increased lethality in young adults occurred because they lacked protective immunity that would be conferred by previous infection with a related virus. Reconstruction of the origins of the 1918 influenza virus provides support for this hypothesis.
Analysis of influenza virus genome sequences using a host-specific molecular clock together with seroarchaeology (analysis of stored sera for the presence of antibodies to influenza virus) indicates that the 1918 H1N1 virus arose ~1915 by reassortment of an avian influenza virus with an H1 virus that had previously emerged around 1907. The 1918 virus acquired the HA gene from the 1907 virus, and the NA gene and internal protein genes from an avian virus. This 1918 virus also infected pigs, in which descendants continue to circulate; however the human 1918 virus was displaced in 1922 by a reassortant with a distinct HA gene.
Seroarchaeology and mortality data indicate that an influenza pandemic in 1889-1893 was caused by an influenza H3N8 virus. This virus appears to have circulated until 1900, when it was replaced by a H1N8 virus (the N8 gene originating from the previously circulating H3N8 virus).
How do these events explain the unusual mortality pattern of the 1918 influenza A virus? High mortality among 20-40 year old adults might have been a consequence of their exposure to the H3N8 virus that circulated from 1889-1900. This infection provided no protection against the 1918 H1N1 virus. Protection of other age groups from lethal infection was likely a consequence of childhood exposure to N1 or H1 containing viruses (this may also have resulted in the lower than usual mortality in the elderly population). Influenza is typically highly lethal in very young children due to lack of immunologic memory.
These observations suggest that childhood exposure to influenza virus is a key predictor of virulence of a pandemic strain. Antibodies against the stalk of the HA protein protect against severe disease, but only within groups of HA subtypes (HA groups are determined by phylogenetic analysis). In 1918, antibodies against a group 2 HA subtype virus (H3) did not protect against severe disease caused by a group 1 HA subtype virus (H1). Childhood exposure might also determine mortality of seasonal influenza. For example, the high virulence of currently circulating H3N2 influenza viruses in those older than 65 years might be a consequence of infection with an H1N1 virus at a young age.
This logic can also explain mortality caused by influenza H5N1 and H7N9 viruses. Most fatalities caused by H5N1 viruses (the H5 is a group 1 HA) have been in individuals who were infected as children with an H3 virus (group 2 HA). Most fatalities caused by H7N9 viruses (group 2 HA) have occurred in individuals who were infected as children with H1N1 or H2N2 viruses (group 1 HA).
The practical consequence of this work are clearly stated by the authors:
Immunization strategies that mimic the apparently powerful lifetime protection afforded by initial childhood exposure might dramatically reduce mortality due to both seasonal and novel IAV strains.
Does this suggest a childhood immunization strategy with a representative mixture of influenza antigen groups, to circumvent or take advantage of original antigenic sin?
Why does the children exposed to influenza virus provide more potent protection than adult exposure?
Yes, that would be one approach. The key would be to induce broadly-protective antibodies to HA stem residues, which confer protection against many strains. That has yet to be achieved.
That first exposure provides immune memory to the infecting virus. Subsequent exposures then produce a memory response to that strain, rather than the infecting strain.
Pingback: Unusual mortality pattern of 1918 influenza A v...
The paper by Worobey et al on the genesis and pathogenesis of the 1918
pandemic H1N1 influenza A virus provides new explanations for the age
distribution of mortality during the 1918 and subsequent influenza pandemics
and following H5N1 and H7N9 influenza (Proc Natl Acad Sci USA 2014 doi/10.1073/pnas.1324197111). However, other factors also affect the pattern of influenza-related mortality.
First, in trying to explain influenza mortality, virologists usually overlook common features of the human host response to influenza and other types of critical illness. Exposure to one influenza virus may affect the probability of infection by another virus years later, but it might not have the same effect on the risk of dying. Influenza mortality isn’t all about the immune response to the virus, and “virulence factors” associated with life-threatening infection often lie within the host. Underlying risk conditions are important; in 1918, a 28 year-old woman who was pregnant had a much higher risk of dying than one who was not. Human evolution also helps explain different mortality rates in children and adults (Antiviral Res 2013; 99: 417-35). A recent report describes a model of low influenza mortality in pre-pubertal mice and much higher mortality after the onset of puberty (Am J Respir Crit Care Med 2013; 187: A1704). The striking mortality difference seen in this model had nothing to do with previous exposure to the influenza virus.
Second, bacterial superinfection is an inadequate explanation for mortality
among young adults in 1918. In 1918, children were infected more frequently
with the same virus as adults and surely had higher nasopharyngeal colonization
rates with the bacteria associated with pneumonia deaths in adults, yet
children seldom died (J Infect Dis 2009; 199: 1408-9). In outbreaks of seasonal
influenza and in pandemics (e.g., pH1N1 in 2009), only about one-third of
patients have had documented bacterial superinfection. Moreover, the median
duration of illness in those who die is about 10 days; they die late in the course of illness when they are immunosuppressed and show signs of a general failure of energy metabolism. Worobey et al suggest “antibiotics and vaccines against several pneumonia-causing bacteria could be expected to reduce mortality dramatically if we were faced today with an otherwise similar set of pandemic ingredients.” For many reasons, a dramatic reduction is unlikely to happen.
Third, Worobey et al state that new influenza vaccines that “mimic the
lifetime protection afforded by initial childhood exposure… (and these
vaccines) … might dramatically reduce mortality due to seasonal and novel IAV
strains.” Although prospect of such vaccines is exciting and they might eventually be developed, it is unlikely they will become globally available in the foreseeable future; in the meantime, a pandemic is much more likely. Experience in 2009 demonstrated the inadequacy of a vaccination approach to managing a
global pandemic. Even in the US, a country better prepared than most,
vaccination affected only 2-4% of all pandemic cases, hospitalizations and
deaths.
Effective management of global pandemic will probably require an
understanding of how to use modern drugs with immunomodulatory activities. For example, observational studies suggest that statins, ACE inhibitors and angiotensin receptor blockers reduce 30-day mortality due to pneumonia and (for statins) influenza. These agents are readily available as affordable generics in every country with a basic health care system, and physicians could use them on the first pandemic day. Unfortunately, influenza scientists and the health officials who support their work (and count on their advice) have shown almost no interest in undertaking research to show whether this approach would work (Clin Infect Dis 2014; 58: 233-7).
theoretically … But was it ever demonstrated in an actual influenza outbreak ? You would assume that by now they had some papers
or experiments. And what about vaccine ? Trivalent vaccine to
children was never questioned because of that OAS effect, afaik.
They did all sorts of crazy theories in 1918 … an association
with infection in 1889-93 -or lask thereof- would have been noticed.
May,28,2010
Vincent Racaniello, PhD, a virologist and Higgins Profession in microbiology and immunology at Columbia University in New York City, told CIDRAP News there is conflicting evidence on the existence of OAS in humans and that it may not be needed to explain the low incidence of pandemic H1N1 in older people. Racaniello is also the author of Virology Blog.
He said several studies have shown the antigenic similarity of the pandemic H1N1 virus and H1N1 strains that circulated from 1918 until the 1940s. Researchers have found that mice that were immunized with a 1918-like vaccine were protected from a lethal challenge with the 2009 virus and that the 1918 and 2009 H1N1 virus hemagglutinins have similar three-dimensional structures, he said.
“In other words, there are ample data which explain the protection of older individuals from infection with the 2009 H1N1 virus as a consequence of the relatedness of the HA [hemagglutinin] of the virus with those that circulated during and after 1918,” Racaniello commented. “There is no reason to invoke original antigenic sin to explain these observations.”
He added that it’s not clear “why one would invoke OAS instead of simply [immune] memory.”
“OAS is of course a form of immune memory in which the memory response is skewed to the original virus, not the antigenically changed one,” he said. “But I don’t think anyone has said that older people preferentially make antibodies against the 1918 virus over the 2009 H1N1, when immunized with the current pandemic vaccine.”
I did not think that this paper gave proper weight to studies in multiple animal models demonstrating increased virulence for this virus. I fully buy into the idea of prior exposure history contributing to the unusual morbidity and mortality curve, there are data suggesting this for the 2009 pandemic virus as well. However, i don’t believe this fully explains the overall increased fatality rate in 1918.
A common response I hear to assertions of increased virulence is that those experiments are done in naive animals, but the same is true for most flu studies using this and other viruses. The claim that the virulence is only due to secondary bacterial infections also doesn’t fully wash for me. I see no reason why secondary bacterial infections should have been more significant in 1918 than during 19th century pandemics, unless the virulence of the virus was such that it caused an unusual degree of damage to the airway mucosa. This seems quite likely to me, as the evidence seems clear that secondary bacterial infections were an important cause of mortality. However, I believe that was largely a function of the virulence of the virus.
My feelings about this paper were that part of it were quite strong, particularly the sections about prior exposure to the 1918 HA or a very similar HA. However, I think the paper, if not the final conclusion, would have been strengthened by collaboration with virologists able to appreciate and account for the literature on the virulence of the 1918 virus. Because the authors didn’t do this, I believe they over-reached.