
 by Gertrud U. Rey
by Gertrud U. Rey
Transmissible spongiform encephalopathies (TSEs) include a variety of fatal neurodegenerative diseases caused by infectious proteins called prions. Although prions are not viruses, their ability to self-propagate without a nucleic acid intermediate has always fascinated virologists, causing them to adopt prions into their repertoire of pathogenic agents. Common TSEs comprise scrapie in sheep, bovine spongiform encephalopathy (“mad cow disease”) in cattle, Creutzfeld-Jakob disease in humans, and chronic wasting disease in deer.
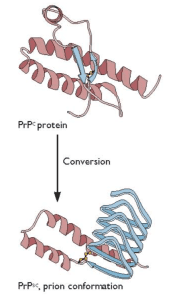
Prions are misfolded versions of a non-pathogenic cellular surface glycoprotein called PrPC (“Prion Protein-cellular”). The misfolded protein PrPSc (“Prion Protein-scrapie”) can adopt different conformational motifs called “strains” and can be acquired accidentally through a medical procedure or by ingestion of meat from an infected animal. PrPC can also misfold spontaneously or due to an inherited genetic mutation. PrPSc is resistant to proteases and acts as a pathogenic, transmissible infectious agent by serving as a structural template to promote the aggregation and conversion of PrPC into PrPSc. A previous post provides a comprehensive review of prions and TSEs.
Sentinel immune cells recognize invading pathogens by sensing structurally conserved molecular motifs known as pathogen-associated molecular patterns (PAMPs) in infectious microbes. This sensing occurs through pattern recognition receptors such as Toll-like receptors (TLRs) on the immune cells. TLR2 in particular recognizes PAMPs produced by bacteria, fungi, or viruses, and this mobilizes innate immune cells such as macrophages, polymorphonuclear neutrophils, and dendritic cells to the site of infection. Pattern recognition receptors also sense host molecules known as damage-associated molecular patterns (DAMPs), which trigger an inflammatory response based on non-infectious “danger” signals. In addition to activating TLRs, PAMPs and DAMPs also activate the complement system. The complement system is a part of the innate immune system that consists of various small proteins that ultimately stimulate phagocytes to eliminate foreign and damaged substances through a cascade of events. The activation of the complement system by PAMPs and DAMPs results in the recruitment and cleavage of complement molecules C3, C4, and C5. Fragments resulting from this cleavage (C3a, C4a, and C5a) generate pro-inflammatory signals by binding to their respective receptors (C3aR1, C4aR1, and C5aR1).
Because the mechanisms that determine neuroinflammation and neurodegeneration during prion infection are unclear, the authors of a recent study sought to determine the function of innate immune elements of the central nervous system during the course of prion disease. The main goal of the study was to establish the relevance of TLR2, C3ar1, and C5aR1 during infection with scrapie strain 22L.
The authors first screened for changes in expression of DAMP receptor genes and other innate immune genes in the brains of mice infected with a mouse-adapted strain of strain 22L. They noted increased expression of complement genes and of both TLR1 and TLR2 in the brains of infected mice, with the gene encoding TLR2 showing the largest increase in expression over the course of infection. The authors also found that while the concentration of C3a or C4a molecules did not change much in the brains of infected mice compared to mock infected mice, the concentration of C5a more than doubled during the course of prion disease, suggesting a possible role for C5a in innate immune signaling.
To assess the effect of individual innate immune receptors on prion pathogenesis in the central nervous system, the authors infected mice deficient in either TLR2, C3aR1, or C5aR1 with strain 22L. Deletion of C3aR1 or C5aR1 had no obvious effect on disease rate; however, mice deficient in TLR2 displayed accelerated pathogenesis compared to control mice. This suggests that a lack of TLR2 signaling increases susceptibility to prion disease and that TLR2 may provide partial protection against disease.
Increasing evidence suggests that the interaction between TLR2 and its ligands plays a role in neurodegenerative diseases. TLR2 is the primary receptor for amyloid beta peptide, which is the main component of amyloid plaques in the brains of Alzheimer’s patients. TLR2 has also been implicated in other neurodegenerative diseases such as Parkinson’s and Lewy body dementia. Furthermore, other studies have shown that TLR4 is also neuroprotective.
These findings indicate that activation of the innate immune system through TLR signaling can interfere with scrapie infection and may have potential positive implications for prion disease therapy. One possible treatment strategy could involve the use of agonists that specifically target TLRs and stimulate host responses to prevent replication of PrPSc. However, this approach requires early diagnosis and treatment of infected patients, which remains a challenge. Nevertheless, we may be one step closer to treating a currently incurable disease.
Pingback: Toll to the Rescue – Virology