
 A sensitive and specific blood test has been developed that could be used to limit the risk of transmission of prion disease through the blood supply (link to papers one and two).
A sensitive and specific blood test has been developed that could be used to limit the risk of transmission of prion disease through the blood supply (link to papers one and two).
Prion diseases, also known as spongiform encephalopathies, are uniformly fatal, chronic degenerative neurological diseases caused by misfolding of a cellular protein, PrPC. Transmissible encephalopathies may be acquired by organ transplant, receiving contaminated blood, or the ingestion of contaminated food.
In the 1990s a new spongiform encephalopathy, variant Creutzfeld-Jakob disease or vCJD, began to appear in Great Britain. Variant Creutzfeld-Jakob disease is caused by prions acquired by the consumption of cattle with bovine spongiform encephalopathy, also a prion disease affectionately known as mad cow disease. To date 231 cases of vCJD have been reported, mainly in the UK and France.
Although the spread of BSE has been controlled by surveillance and feeding restrictions, it is estimated that millions of people were exposed to BSE prions. The concern is that some of these individuals might be infected but show no symptoms of disease. If they donate blood, they may transmit infection to others. It is known that several cases of vCJD have been transmitted from infected blood donors, so further transmission is a major concern. So far prion diseases have only been diagnosed after death, by detection of conformationally altered prion proteins in the brain.
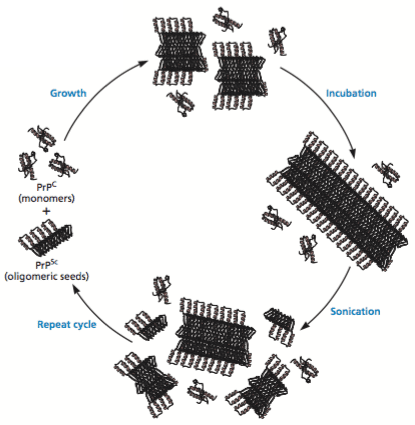
Two sensitive and specific assays for vCJD prions have now been developed that show promise for non-invasive pre-symptomatic diagnosis of the disease. They are both based on a technology called protein misfolding cyclic amplification (PMCA, illustrated; image copyright ASM Press, 2015). A small amount of the normal human prion protein, PrPC (produced in transgenic mice) is mixed with plasma. The samples are incubated to allow formation of prion oligomers, followed by disruption by a pulse of sonication to disrupt the oligomers. The cycle is repeated multiple times, much like polymerase chain reaction (PCR) which is used to amplify small amounts of DNA. Prions are detected by western blot analysis after treatment with proteinase K. The misfolded, pathogenic prions, PrPSC , are not completely digested with this enzyme.
In one study, PMCA was used to analyze blood samples from 14 cases of vCJD and 153 controls, which included healthy individuals and those with other neurological diseases, including sporadic CJD (sCJD – not caused by ingestion of contaminated beef). All 14 samples from cases of vCJD were positive in the PMCA assay, but not any of the other samples.
In a second study, the PMCA assay was positive in samples from all 18 patients with vCJD. Of 134 control samples, just one was positive for vCDJ, from a patient with sCJD. Furthermore, the assay detected vCJD prions in archived blood samples from donors who gave blood before developing symptoms of the disease.
These findings suggest that the new assays can detect vCJD prions in the blood before the appearance of the neurological symptoms of spongiform encephalopathy. While additional samples must be analyzed to validate the results, they are nonetheless promising as a way to prevent spread of the disease via the blood supply. Unfortunately, if you are diagnosed with vCJD by one of these assays, that is the only positive outcome – there are as yet no treatments for any spongiform encephalopathy.
Pingback: Prions in bacteria
Can I obtain a blood test for suspected vCJD or Prion disease.. I have horrendous symptoms and am extremely sick.